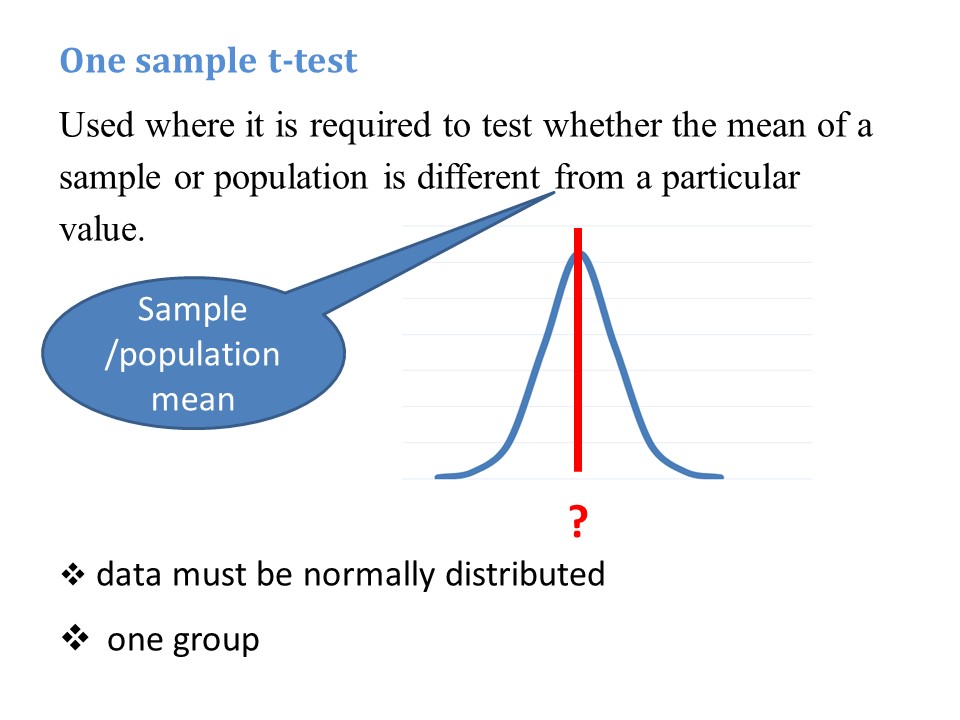
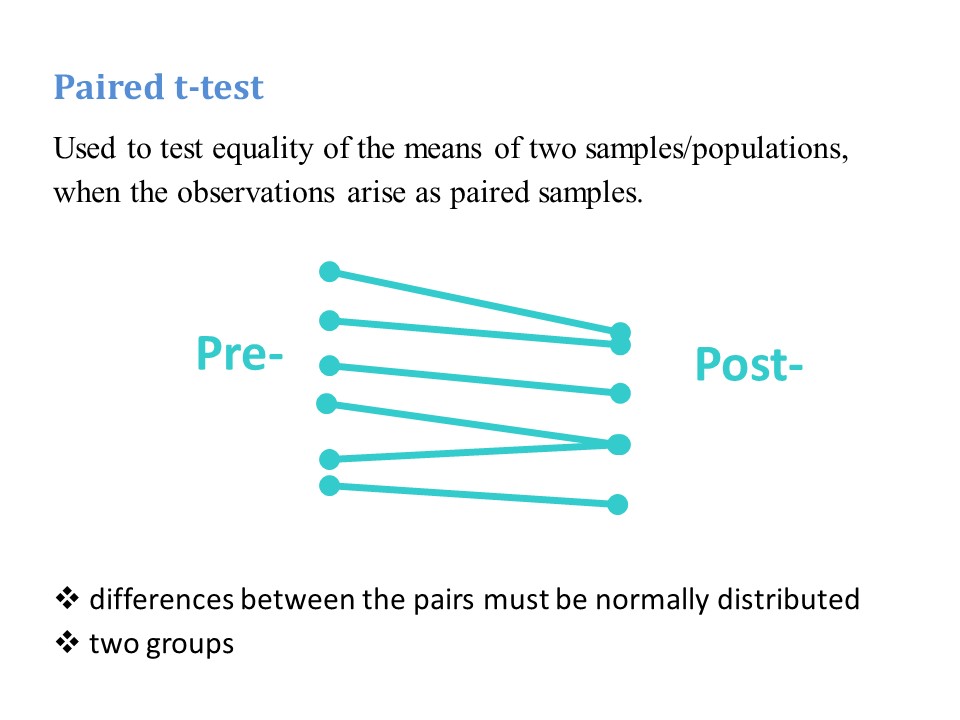
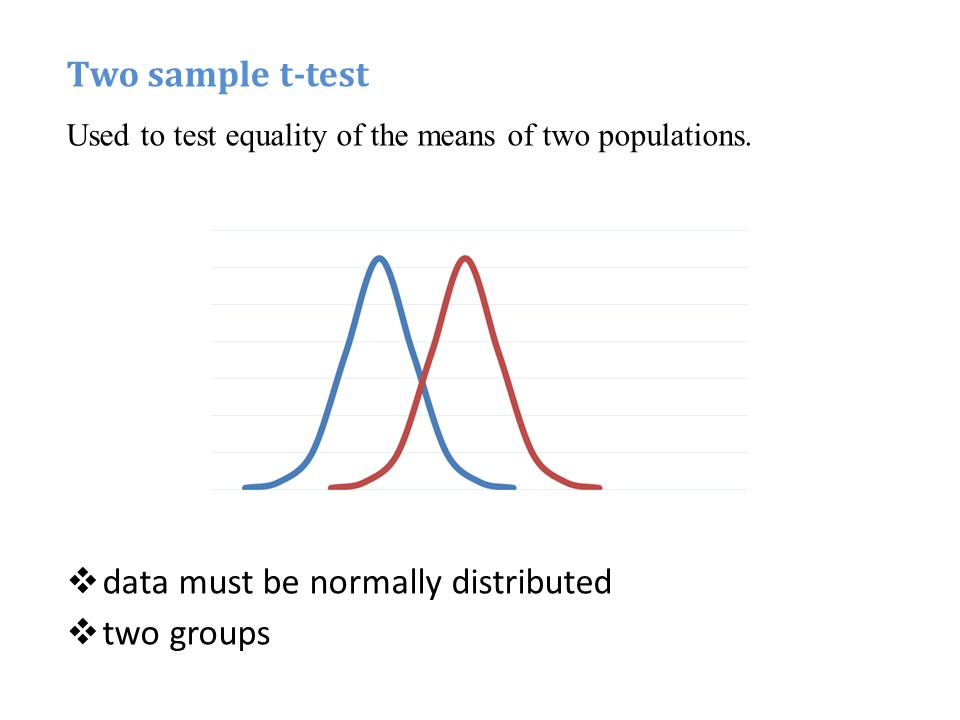
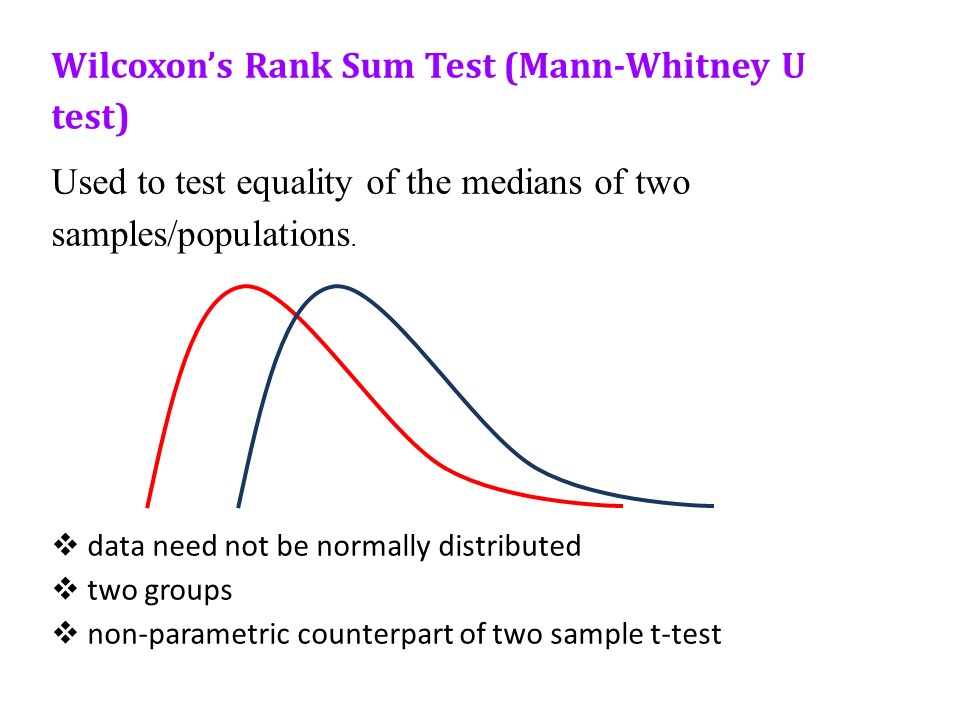
These remarks apply to both a ‘standard’ negative binomial regression and a ‘zero truncated’ negative binomial regression. A zero truncated negative binomial regression is appropriate when there are no zero values, for example if you are counting days in hospital. In the current example, our outcome variable is number of hours on a ventilator. Explanatory variables are Apache Score (a measure of disease severity) and sex.
The negative binomial regression model will output either a standard set of coefficients or an exponentiated set of coefficients, which reflect the IRR. You will see a coefficient for each of the explanatory variables in the model, and a coefficient for the constant term.
The IRR is the exponential of the coefficient, not the log of the coefficient. Remember it is NOT a linear regression, but a (zero truncated) negative binomial regression. Two different kinds of regression.
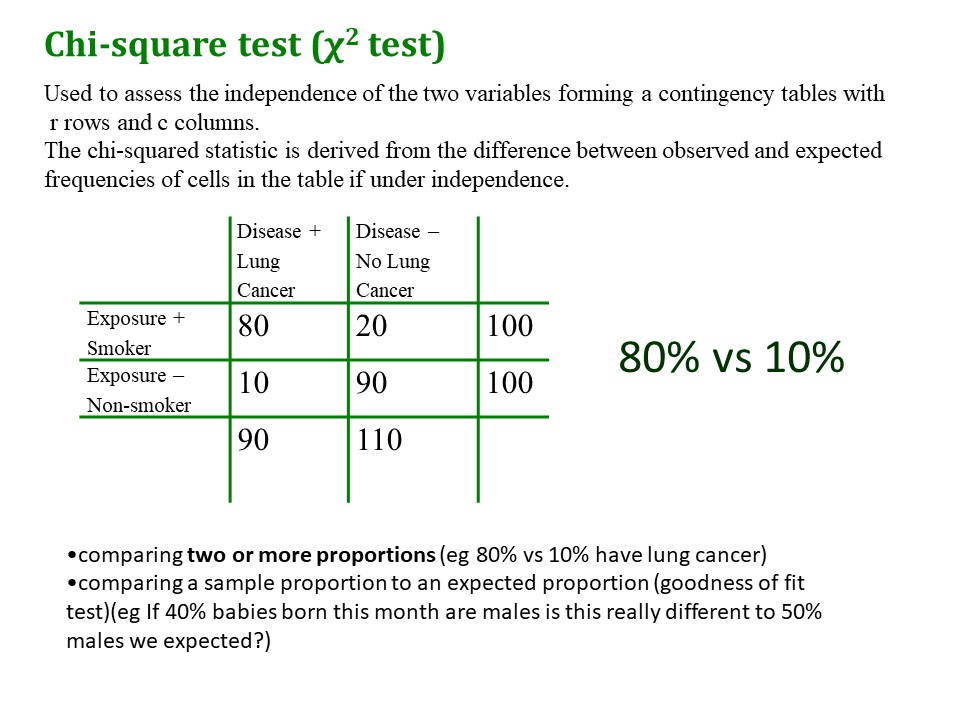
Incidence rate ratio: the rate
The ‘rate’ part comes in because the outcome variable is actually a count, in this case a count of ventilation hours. We really mean it is a count of ventilation hours per person.
Contrast this with another scenario where you are looking at aircraft flight delays over a number of different airlines. You can’t just talk about the number (count) of flight delays, you have to take into account the number of flights a particular airline may have scheduled. Five flight delays out of ten scheduled flights would be terrible, whereas five flight delays out of 500 flights would not be too bad. With this kind of data you enter the ‘exposure’ variable (number of scheduled flights) as what is called an ‘offset’ variable into the regression equation.
But in our current example, we are talking about ventilation hours per one person, so we don’t have to include an offset.
to see how the ratio part works. Here I used an example where we had ventilation hours as the outcome variable with two predictors, apache3 (Apache III score, a numeric indication of the severity of illness, higher is worse) and sex (male=0, female=1). The IRR for apache3 is 1.0035. This is equal to e^0.0035, the coefficient for the neg bin equation.
The IRR is the ratio of the ventilation hours at a given level of apache3 (say 101) divided by the predicted ventilation hours at an apache3 score of 1 unit less (say 100), holding sex constant. It’s easier to see this if you look at row 15 of the spreadsheet – at apache3=101, ventilation hours = 50.07, at apache3=100, ventilation hours = 49.9.
50.07/49.9 = 1.0035, the ‘incidence ratio’ of ‘new’ hours on a ventilator. You can see this holds true for any value of ventilation hours.
Have a play with the spreadsheet inputting different values for apache3 and sex. You could use this with your own equations to see the predicted outcome measure of your own interest.
Why write a paper? You would have started with an idea, developed a hypothesis, designed a study, and conducted your study to prove or disprove your hypothesis. Now you need to disseminate the results. ‘If it is not published it never happened’.
This post may help you get organised to write your paper. A pdf can be downloaded: How to write a paper
Getting started
To get started, you should:
Tell a story – engage the reader’s attention
Start writing as soon as you have the idea
Write within a structured design (IMRAD)
READ THE INSTRUCTIONS TO AUTHORS for the journal of your choice.
Put headings in first, then fill sections
Before you start, assemble and READ the following documents:
your study protocol
your ethics application and approval
the appropriate reporting guideline (eg CONSORT, STROBE, STARD, ARRIVE), download from the equator network page http://www.equator-network.org/reporting-guidelines/. Note that all the guidelines have some sort of companion document which explains and gives examples of all the items.
the SAMPL guideline for statistical reporting, also from the equator network
the instructions to authors from the journal you have selected for submission.
Resources for writing your paper include your original study protocol, your approved application for ethics approval if relevant. These documents will contain much of the information you need. If these are well written all that might remain is for you to fill in the results and write your discussion! These steps can be envisioned as a cycle.
The final step is to submit your manuscript to the journal of your choice.
Structure of a scientific manuscript
Your manuscript will include a title page, abstract, body of the manuscript, references and acknowledgements. The recommendations from the International Committee of Medical Journal Editors are particularly useful when embarking on writing your paper: http://www.icmje.org/recommendations/browse/manuscript-preparation/preparing-for-submission.html#d
Title page
Title
The title should be:
Concise and descriptive
Convey essential features of the article’s content
Include key words to be picked up by electronic search
For example, the following publications give a good sense of what the manuscript is about:
The medical management of missed miscarriage: outcomes from a prospective, single-centre, Australian cohort. Petersen SG, Perkins AR, Gibbons KS, Bertolone JI, Mahomed K. Med J Aust. 2013 Sep 2;199(5):341-6.
Serum vitamin D levels are lower in Australian children and adolescents with type 1 diabetes than in children without diabetes. Greer RM, Portelli SL, Hung BS, Cleghorn GJ, McMahon SK, Batch JA, Conwell LS. Pediatr Diabetes. 2013 Feb;14(1):31-41
Authors
Decide on the authors and order of authors BEFORE you begin any research study. This prevents misunderstandings and possibly arguments later. In the biomedical sciences, the person who did research and wrote the paper is conventionally the first author, the head or leader of the research group is last, and everyone else goes from first to middle or last to middle in decreasing order of importance/contribution.
Authorship must be justified. Authors must have contributed intellectually and substantially to the research and the manuscript. People who helped but did not contribute intellectually, for example technical people, may be included in ‘acknowledgements’ at the end of the paper
Use both or all initials of authors, to facilitate electronic searching. Make sure you have the correct qualifications, affiliation/s, and email address for each author.
Abstract
The abstract should follow the IMRAD format, as for the main body of the text. Features of the abstract:
200 – 300 words
Check instructions to authors!
Will be used for electronic searching
Often the ONLY report of a study which is read.
Body of the Manuscript
The body of the manuscript should be structured around the IMRAD format:
I Introduction
M Methods
R Results
A and
D Discussion and Conclusions
Introduction and background
The introduction should be short, usually just a few paragraphs. Avoid a detailed discussion of the literature – this comes in the discussion section. You only need to convey the following points which should be addressed:
Why did you undertake your study? Relate your reasons to the clinical or research question
Why is your study better than previous reported studies? Describe any problems, limitations, or gaps in knowledge from previous work
Use only essential references
Be specific when citing previous work –readers find it helpful if you specify the author/s, subject numbers, year of publication and effect size. For example
‘Smith et al, in their 1965 study of 2000 mice, found that black mice had on average 20% (95% confidence interval 18-22%) higher problem solving ability than white mice’…..gives more complete information than ‘a study found that different coloured mice had different problem solving ability’….
The AIMS of the study are described at the end of the introduction, as well as specific hypotheses to be statistically tested.
Methods
The purpose of the ‘methods’ section in your manuscript is to enable the reader to understand what you have done, and to be able to reproduce your results.
The various standards of reporting are invaluable guides to structuring your methods section. Each standard, guideline or checklist has a list of items which should be addressed in the methods section.
The following standards are commonly applicable:
Randomised controlled trials – CONSORT
Observational (cross sectional, cohort or case control) studies STROBE
Studies of diagnostic tests STARD
Studies involving animals ARRIVE
There are many other checklists available, for a comprehensive list as well as those above, see the Equator Network website http://www.equator-network.org/. As well as items in the appropriate checklist, make sure your methods section reports on:
Ethics and governance
Bias (remember the main types are selection, misclassification and confounding).
Describe the methods used for each aim/hypothesis in the same order as given in the introduction.
Results
In the results section you:
Report what was found – in the first paragraph; make sure this corresponds to the aims and hypotheses stated at the end of the introduction.
Describe participants/study units
Describe in detail the ‘answers’ to main question/s in same order as in methods
Write on one topic per paragraph
Don’t duplicate information in text and tables/figures. The results section is often quite short – a few paragraphs.
Tips:
Give numerical results to appropriate decimal place
Report numerator & denominator as well as percentage
Give exact p-values, don’t use ‘NS’ for not significant
Give p-values to appropriate decimal place – 0.64, 0.03, 0.003, 0.0005, <0.0001 (again, check the instructions to authors – some journals specify the use of <0.001 if the p value runs to four decimal places)
Report the mean with standard deviation or mean with 95% confidence intervals for normally distributed data. Read the instructions to authors, many journals specify which summary statistic they prefer
Report the median with interquartile range for skewed data (in some circumstances it is appropriate to also report the range).
To ensure that your statistical results are reported correctly, refer to the SAMPL guidelines, which can be downloaded from the equator network at http://www.equator-network.org/reporting-guidelines/sampl/.
Discussion and conclusion
The first paragraph should contain a brief summary of your main findings, without re-stating the results too much. Don’t forget these conclusions should correspond to, and be in the same order as, your original aims and objectives. i.e. what was the ‘answer’ to your research questions/s?
Discuss each result in the same order as you reported aims, methods, and results, then give a global overview of the impact and meaning of your work in the context of current knowledge.
In discussing each result, give your interpretation of the meaning or implications of your work. Refer to previous work and explain whether your work agrees/supports or does not support previous results. Explain how your work expands on previous knowledge. When citing previous publications, mention any limitations or biases as well as strengths of previous work, i.e. do a mini critical appraisal. When describing results of your own or other author’s work, be as specific as you can, giving number of subjects and effect sizes rather than general statements. For example, rather than saying ‘caesarean section rates have risen’, say ‘Smith and Jones, in their UK study of 150,000 pregnancies from 1990 to 2010, found that the annual caesarean rate rose from 20% (95% CI 18 – 22%) to 35% (95% CI 33% – 37%)’.
Explain the limitations of your study. A good place to start is consideration of any biases – selection, misclassification or confounding biases. Is your study sample representative of the population you are interested in? Did you ‘cherry-pick’ the data in any way? Discuss the generalisability (external validity) of your study, and whether it was study was adequately powered to find a positive result.
In conclusion, re-state the ‘answers’ to your research question/s and take-away message. Do NOT state that ‘further research is required’! If you really must recommend further research, outline a specific study design and plan, and put it before the conclusions towards the end of the discussion.
Writing tips and the final check
Put the manuscript away for a day or two then re-read
Use short sentences
Prune ruthlessly – ask yourself, can I say this in fewer words? Space in journals is at a premium; for both journal articles and longer documents such as theses, succinct phraseology enhances readability
Delete extraneous words ‘such that’, ‘however’, ‘significantly’ (you have already defined statistical significance in the methods); distinguish between statistical significance and clinical or scientific importance
Give informative citations – consider including author, year, number, effect size
Get an educated ‘lay’ person to read your manuscript; if they can’t understand it, revise!
Do a final proof read to ensure that all numbers are consistent and add up appropriately, and that all percentages reported contain both numerator and denominator
Double check the instructions to authors and make sure all the formatting is correct, the word limit is adhered to, and any other conditions are complied with.
Submitting your manuscript
Choose a journal. Consider the readership, the journal’s impact factor, and the purpose of journal
READ THE INSTRUCTIONS TO AUTHORS
Make sure the references are cited correctly for that journal
Write a letter to editor saying what is novel and important about your paper and how it adds to current knowledge
Most journals require electronic submission. If the receiving editor decides your manuscript is good enough and meets the journal’s policies, it will be sent out for review – usually two or three reviewers. The reviewers will make recommendations, which might be:
Accept – usually with minor or major revision or
Reject
You will be notified by the editor of the decision. The reviewer’s comments, which will be sent to you, are invaluable! If your manuscript is accepted subject to revisions, it is very important to address each comment in detail and with professional courtesy and respect to the reviewer. Always thank them for their comments.
It is hard to get published in a high-ranking journal! For example, for Thorax ( http://thorax.bmj.com), published by the British Thoracic Society and British Medical Journal group, the ‘instant rejection rate’ is 66%, and the acceptance rate 8% at the time of writing (November 2017).
Here we’ll take a brief, but hopefully practical, overview of sampling in the context of designing a research project, and how to select a suitable sample size.
The participants you include in your study are an important consideration, to ensure that the results of your study can be extended or generalised to a larger group than your actual study participants. This is known as ‘generalisability’, or ‘external validity’.
Sample size is important to make sure that you include enough subjects to have a reasonable chance of detecting a difference between groups, for example difference in response rates to a new treatment compared with an old treatment, if there is one, and that you do not unnecessarily include too many subjects which might be unethical or an unjustifiable use of scarce research funding or resources. Having said that, a larger group is usually very desirable to improve the precision of the findings of your study and enable more detailed analyses to be run.
Study protocols and ethics applications invariably require a justification for sample size.
What is a sample?
If we want to know the exact ‘truth’ of something, we can measure every person (or item) in a whole population. This is a ‘census’ – ‘a study that involves the observation of every member of a population’. Measuring an entire population, for example in the periodic Australian census, is expensive and inconvenient, and not usually feasible in the context of medical research.
Instead we select a much smaller number of subjects who we hope are representative of the population we’d like to study.
We assume that this reflects the population, and that the results are estimates of the ‘truth’. Hence, summary statistics such as mean, median or proportion are given with standard deviation, standard error or a confidence interval, to indicate the precision of our estimate.1
We do a research study, in the biomedical or veterinary context, in order to apply the result to some ‘target population’. For example, if you are a respiratory clinician, you might like to know whether dornase alfa administered to children and adolescents aged 5 – 18 with cystic fibrosis was effective in improving lung function in this group.
The target population in this example is ‘children and adolescents aged 5 – 18 with cystic fibrosis’. It is self-evident that it would not be feasible to test the drug in ALL people with cystic fibrosis aged 5 – 18 in the world; however we would like to apply the results to this group in general. Thus, the research needs to be conducted in a group which represents all people with cystic fibrosis aged 5 – 18 years.
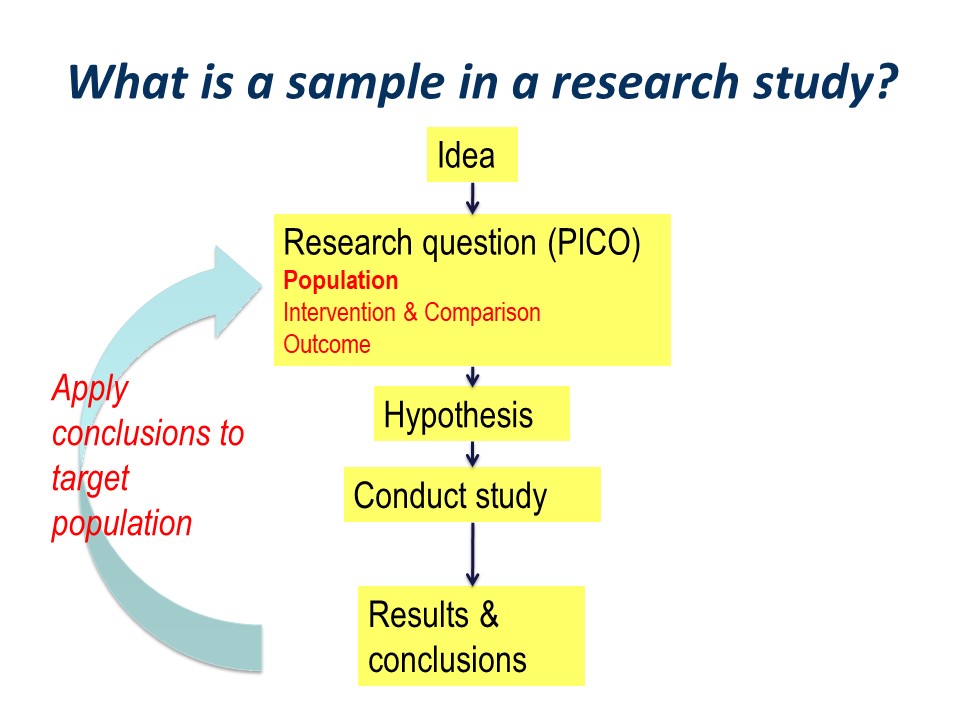
The research process can be pictured as an idea, followed by development of a structured research question, typically along the PICO guideline of Population, Intervention, Comparator. This is followed by development of specific statistically tested hypothesis or hypotheses. We then go ahead and conduct out study, think about our results and draw our conclusions.
Figure 1. The research cycle from the idea to application.
Hopefully, the conclusions will be applied to our original wider population of interest. This is the essence of the process of translation of research into practice.
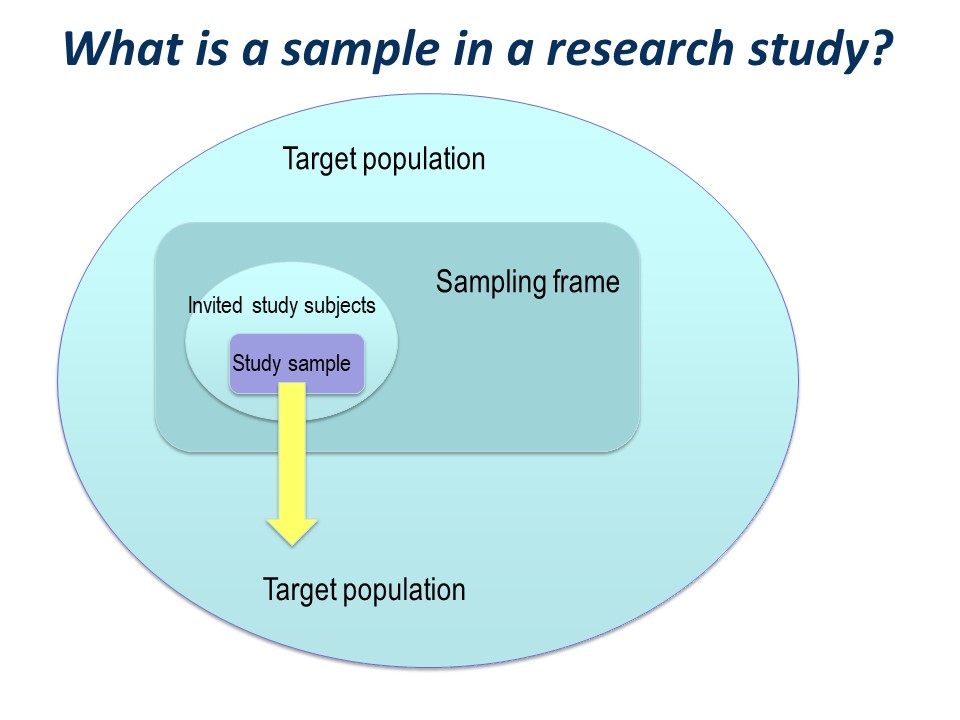
There is a big ‘step down’ from the target population to the actual study sample. It is very useful to conceptualise this using a diagram like this on – you can see how important the selection of the study sample is.
You have a large ‘target population’, the population to which you wish to apply the conclusions of your study.
Everitt defines a target population as
‘The collection of individuals, items, measurements, etc. about which is is required to make inferences. Often the population actually sampled differs from the target population, and this may result in misleading conclusions being made’.1
The sampling frame
Then you have a ‘sampling frame’. This is the group of subjects to which you have access – essentially a list – from which you hope to select your study sample.
It could be quite specific, for example, you may have an actual list of all the patients in your clinic. You may have a list for potential control subjects.
It might be less specific; you may not have a list of patients in your clinic, but plan just to invite everyone who comes to clinic over a specified time period, say 3 months or 6 months.
It may not be practical to invite every single person in your sampling frame to participate in the study. For example, if you are recruiting over a 3 month time period, those attending over the other nine months of the year will not be invited. Thus, not everyone in the sampling frame is likely to be invited to participate in your study.
You might have a list of 6,000 people from whom to select your study subjects – it is not practical to invite all of them to participate in your study.
So the invited study subjects are a subset of the sampling frame. (See selecting a random sample in later slides).
The study sample
Of those who are invited to participate, not all will agree or consent to being part of your study. So in turn, the initial study sample will be a subset of the invited subjects.
Finally, of those people who consent to participate in your study, some may prove ineligible for the study, if there is any kind of screening for eligibility, and some are likely to drop out.
The subjects left at the end of the study, for whom outcome information is available, constitute the final study sample.
Figure 2. Target population to study sample.
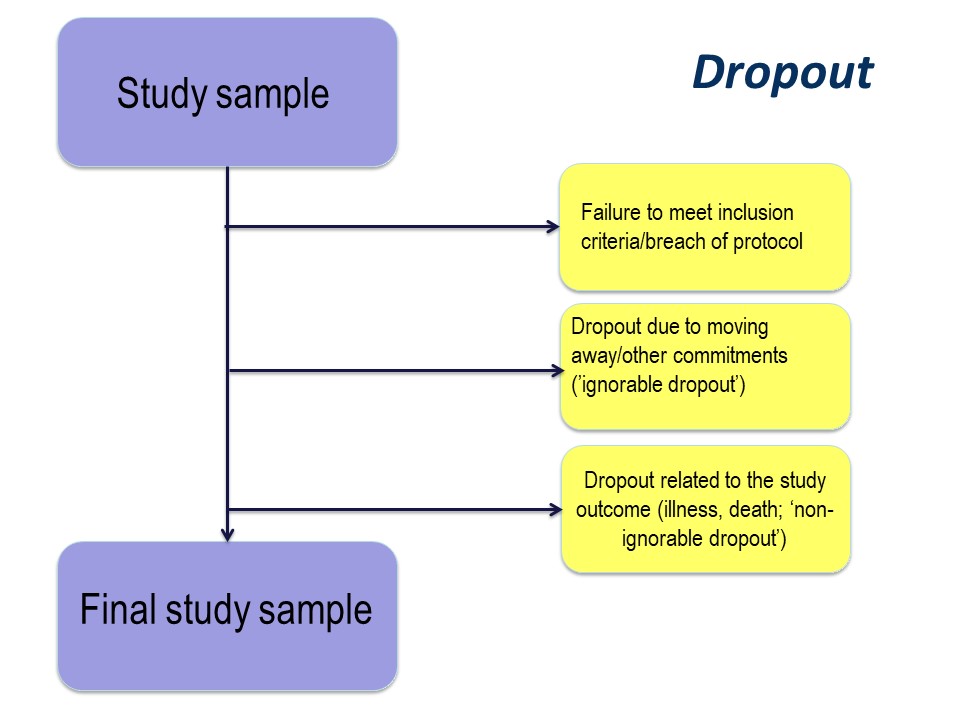
Dropout
The reason for drop out is important. Participants may drop out of studies because of simply electing not to continue with a study, by moving away, or because of illness or death.
Any study dropout is undesirable, but drop out because of illness or death must be taken into account in the statistical analysis. For example, in our cystic fibrosis example, the study was conducted over 96 month. If, hypothetically, some had dropped out because of illness (with very poor lung function) or death, especially if there were more dropouts in the active treatment group, those surviving until the end of the study will be those with the best lung function. This might produce a spurious ‘result’ of improved lung function in the treatment group.
The importance of the reason for dropout will vary depending on the nature of the research that you are doing, but it is very important to consider the implications of dropout at the study design stage.
Figure 3. Dropout from the initial study sample.
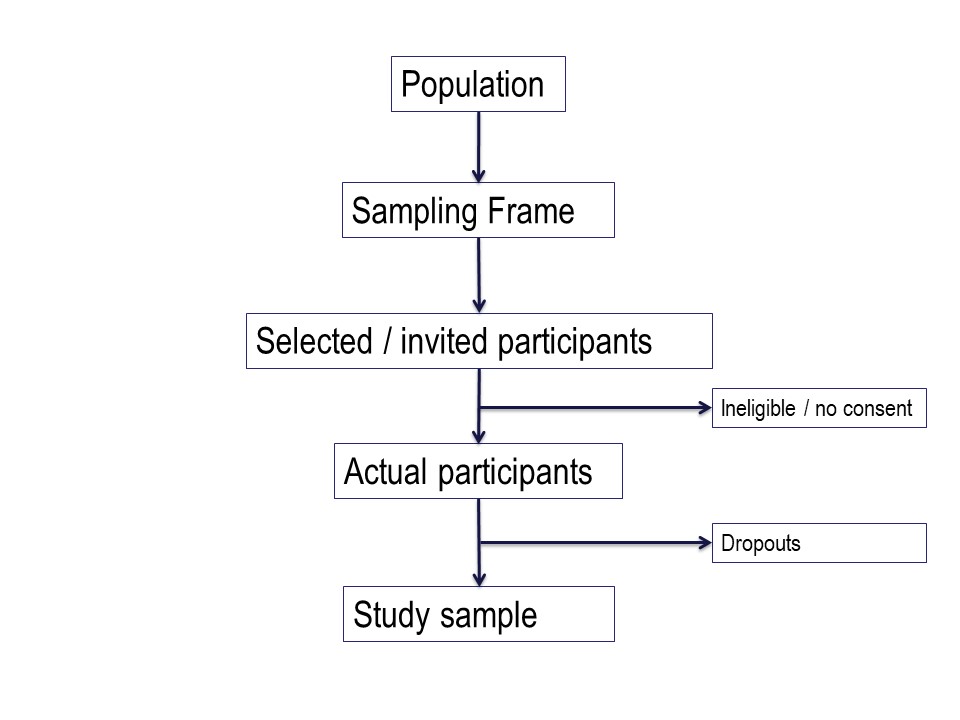
Summary and practical example – target population to study sample
Figure 4 below is a summary of the population to which the study results are intended to be applied, and the flow of patients through a study. Note that essentially all of the ‘standards of reporting’ such as CONSORT .2and STROBE,3 by which editors and reviewers are guided, strongly recommend the use of a flowchart in your manuscript. If you are writing a Cochrane Review a flowchart is essential.4
Figure 4. Selection process through a study.
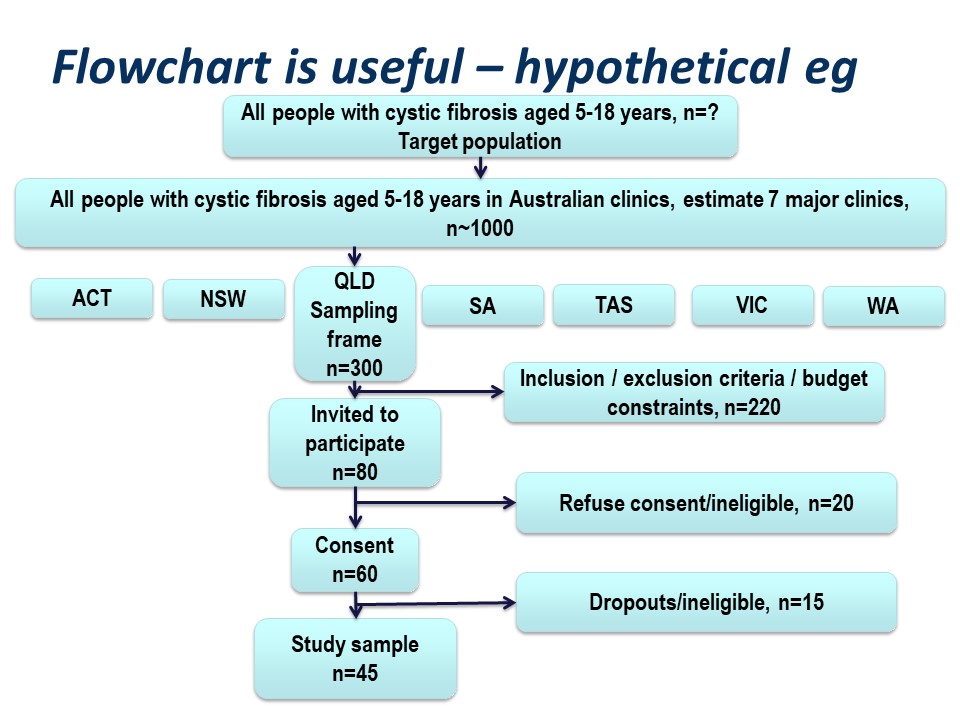
Hypothetically, suppose we want to run a trial of some drug, or an observational study, in people with cystic fibrosis. We want our conclusions to be applicable to all people with cystic fibrosis. We are based in Queensland and have access to patients of the Queensland clinic. Are our potential study subjects representative of all people with CF?
Consider the flow of subjects through the study. We are restricted by feasibility to the Queensland clinics, probably restricted to the Brisbane clinic. This might give us a sampling frame of 300 people. From this, we consider the inclusion and exclusion criteria, which will render a proportion of subjects ineligible for the study at this step. Additionally, we may have budgetary constraints that limits the number of subjects we can include in the study. We have to ‘guestimate’ the likely study sample, the potential dropouts to the study, and work backwards to invite a suitable number of participants. Let’s say we invite 80 people to participate.
Of those invited to participate, not all will consent. Some patients will be found to be ineligible during the patient information step of obtaining informed consent. Here, we have 20 patients who either refuse consent or are ineligible when we initially invite them to participate.
Additionally, sometimes it is not possible to assess subjects for eligibility before consent, for example if some screening test, like a blood test, needs to be applied. In this hypothetical example, we lost some subjects who are ineligible at this point, and others who may drop out for reasons either unrelated or related to the study. We lose another 15 subjects here.
Thus our final study sample is 45 people (Figure 5).
Figure 5. Flowchart for a hypothetical study in cystic fibrosis.
What sample size do I need to select?
This brings us nicely to the topic of sample size – ‘how many subjects do we need in our study’, because the statistical analysis will be performed on the study sample, i.e. those subjects who have actually participated in the study.
Estimates and measures of uncertainty
To expand a little on estimates and confidence intervals:
Any statistic (mean, median, proportion) calculated from the study sample is only an estimate of the ‘true’ statistic we would get if we measured every person in the target population
If we started again and selected a different sample, we would get a slightly different statistic. For this reason, statistics calculated from samples should be reported with a confidence interval. For a 95% confidence interval – if you repeated your study 100 times, 95 times out of 100 the statistic would lie within the confidence intervals.5-7
For example for 20 subjects, the mean FEV1 and 95% confidence interval might be 60% – 80%. As the number of subjects increases the confidence interval decreases. If you measured 40 subjects, the mean FEV1 might still be 70% or something close to it, but the confidence interval would be narrower, say 65% – 75%.
Figure 6. The confidence interval narrows (estimate becomes more precise) as the number of study subjects increases.
Clustering
Valid conclusions assume that the study sample constitutes a random sample. Everitt1 defines a random sample as:
‘….a sample of n individuals selected from a population in such a say that each sample of the same size is equally likely’. 1
For example, in the diagram, if the blue circle represents the sampling frame, and the red dots represent subjects in your study, your comparisons assume the first picture, not the second.
Figure 7. Random sample and clustered sample
If there is clustering:
Firstly, the results will be biased – because the sample is not representative. You may have selected subjects who are sicker, older, younger, richer, poorer, or different in some other attribute, than the average in the target population. Similar comments apply to any control or reference group.
OR it may be possible to adjust for clustering in analysis – you can use techniques like stratifying your study subjects, or more sophisticated techniques which are outside the scope of this article.
Practical calculation of sample size
Calculating sample size is a whole field of study in itself, but in its basic form is easy. For example, in a cross sectional study, you might wish to measure the proportion of mothers at a particular hospital who deliver by caesarean section. How many mothers do you need to assess for mode of delivery to obtain a reasonably accurate or precise estimate?
You might want to know if term babies born by caesarean section are of ‘normal’ mean birth-weight. You may already know what a normal birth-weight is for Australian born babies.
Probably the most common type of sample size calculation is for a comparison of two means. Most sample size calculations come down to two main types:
Planning your study: sample size questions
How many subjects do I need to show a statistically significant effect (if there is one) of my intervention/risk factor? An alternate way of phrasing this question is ‘how many subjects to I need to show a clinically significant difference of X between two groups?’ – in this latter example you need to decide yourself what you regard as clinically important, for example you may decide that an average in birth-weight between two groups of 100g is clinically significant.
Resources for calculating sample size (or power): online calculators and statistics software
There are many on-line calculators for sample size. Two good ones are:
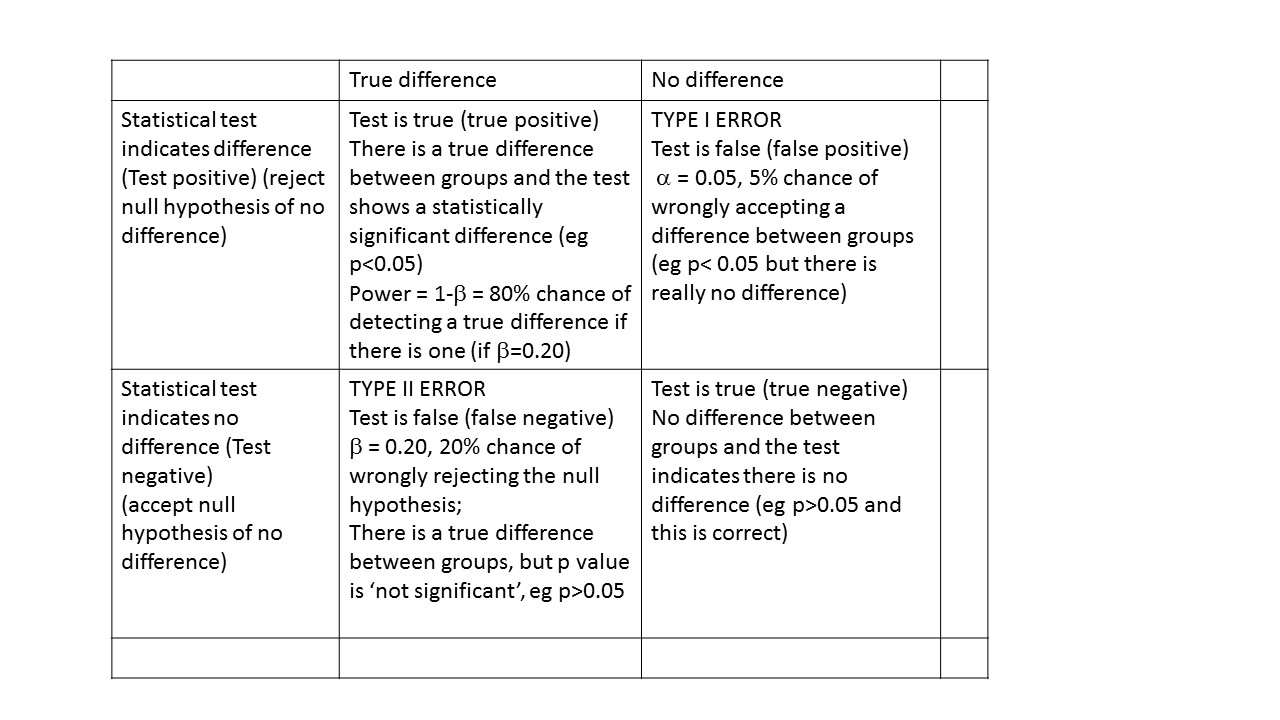
Statistical notation often uses greek letters. Mean is often represented mu (m) and standard as sigma (s). The Type I error is represented as alpha (α) and Type II error as beta (b).
Sample size for prevalence (proportion)
You will need to ‘guess’ the expected prevalence, based perhaps on prior studies or pilot studies, and you need to specify how precise you want your estimates in terms of precision and confidence interval. Typically researchers use 5% precision and 95% confidence interval.
Prevalence – expected prevalence from literature or previous experience
Precision – how accurately you wish to measure your prevalence
Confidence interval – (1 – precision)
Population size – if unknown estimate will not be adjusted for small population size
We will use a hypothetical example where we wish to estimate the proportion of women in a particular hospital who deliver by caesarean section. We think, from our clinical experience, that around 27% of women delivery by caesarean section. We decide on 5% precision and 95% confidence interval.
In the ‘sampsize’ web calculator (http://sampsize.sourceforge.net/iface/), enter the desired precision, prevalence and confidence level, click on ‘calculate’ and your sample size will come up. The calculation shows that we need to ascertain the mode of delivery in 303 women.
Note: the website specifies that if the prevalence is unknown, enter 50%. This is because measurement of a prevalence of 50% requires the largest sample size, so is a very conservative or ‘safe’ estimate of sample size.
Sample size for two means or two proportions
You might want to calculate a sample size for comparing two means, or two proportions.
For example, you might want to compare the mean fetal weight of infants of women with excessive gestational weight gain with those with acceptable gestational weight gain (this example is adapted from Walsh8).
You might want to compare the proportion of women in Australia having caesarean section with the proportion of women in England having caesarean section (adapted from Prosser9).
Power and sample size – Type I and Type II error
You need to consider the acceptable level of type 1 or type II error.
The type I or alpha error is often set at 0.05. This is the probability of wrongly accepting a difference if there is really no difference between groups. The ‘alpha’ is the p-value which is the researcher chooses at the beginning of the study, at the design stage. There are many types of statistical tests, all of which result in a ‘test statistic’. For example, a t-test results in a ‘t’ statistic; a chi-square test results in a ‘Chi-square’ statistic; an analysis of variance (ANOVA) results in an ‘F’ statistic.
The p-value is the probability of obtaining a statistic as extreme, or more extreme, than the test statistic, whatever that test statistic might be. In practice, this is the value on the x-axis of a distribution.
For example, the probability of obtaining a t statistic equal to, or more extreme, than plus or minus 2.042, (with 30 degrees of freedom), is 5% or less. The p-value would be 0.05 or less. Look at a table of Student’s t-distribution in any statistics text book to see how this works. Conversely, the ‘critical value’ of t if alpha is set at p-0.05 is plus or minus 2.042.
[Image to be inserted]
The ‘power’ of a study is the chance of detecting a difference between groups, if there is in fact a difference. For biological studies, power is often set at a minimum of 80%, i.e. the study has an 80% chance of detecting a difference between two groups. Power is related to a quantity called ‘beta’. Beta is often set at 0.20 or 20%. Power is calculated as:
power = 1- β
so if beta is set at 0.2, power is correspondingly equal to 0.8 or 80%. This is probability of falsely rejecting the alternate hypothesis, i.e. concluding there is NO difference between groups when there is in fact a difference. An under-powered study might find no difference between groups when in fact there IS a difference.
For example, if we found that estimated fetal weight at 34 weeks was 2681 g compared with 2574 g, and we conclude this is different, but in fact is due to chance (sampling variation), this would be a type I error. At an pre-set alpha of 0.05, there is a 5% chance this will happen.
Type I & Type II error
For example, if we found that estimated fetal weight was 2681 g compared 2574 g and we conclude this is not different, but in fact really is different, this would be a type II or beta error.
Power can be interpreted as the probability of finding a difference between groups, if there is one.
Sample size for two means
To calculate the sample size for comparing two means you need:
Your estimate of mean 1 (μ1) and mean 2 (μ2)
Your estimate of the standard deviation (SD) of the means
Your decision about alpha
Your decision about beta
The difference between means is known as the ‘effect size’. If the means are close together, as in the picture on the left, you will need many subjects. If the means are far apart, with a large effect size, you will not need so many study subjects.
We will use the stat.ubc website (www.stat.ubc.ca/~rollin/stats/ssize) to calculate a sample size to detect the difference between two means. You need to know the power you want your study to have, typically 0.8 (80%), the alpha value or p-value, typically 0.05, and ‘guestimate’ of the pooled standard deviation of your two groups.
Notice you can solve for either power or sample size
Enter mean one and mean two (often known as mu 1 and mu2)
Enter values for alpha and power
Click on calculate.
For this example, I used the fetal weights and standard deviations from Jennifer Walsh’s paper.8 If mean 1 is 2681 grams, mean 2 is 2574 grams, with standard deviation 345 (standard deviation is often represented by the greek letter sigma), you can see the sample size needed is 164 in each group.
If you want a greater than 80% chance of detecting a difference if there is one, you can increase the power of your study. Similarly if you want a smaller chance of falsely concluding there is a difference when there is really none, you can decrease you alpha value.
Try this with power of 90% and alpha of 0.01!
What was the power of the study actually conducted by Walsh?
Sample size for two proportions
To calculate the sample size for comparing two proportions you need:-
Your estimate of proportion 1 and proportion 2 (p1 and p2)
Your decision about alpha
Your decision about beta
Again using the ‘stat.ubc’ website (www.stat.ubc.ca/~rollin/stats/ssize), we enter our data. For this example, data from Prosser’s paper comparing caesarean rates in Queensland and England9 was used. To detect a difference in a proportion of 0.36 compared with 0.25, we need 274 subjects in each group. Note that the ‘real’ report includes many more subjects and analysed many more variables than merely the proportions of women having cs.
Other sample size calculations
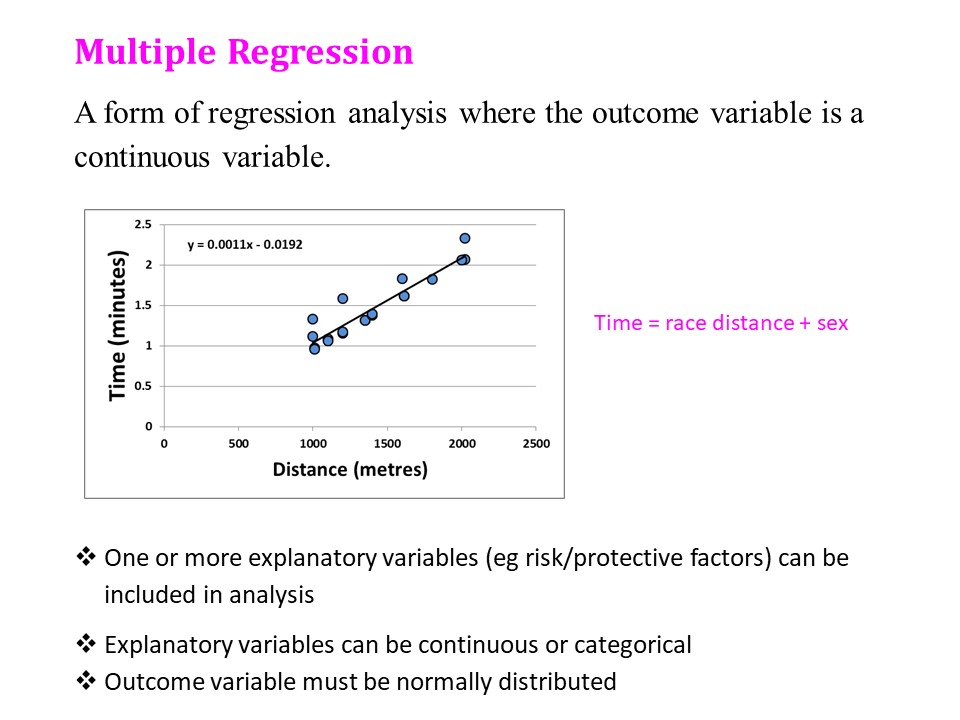
There are many more situations where a calculation for power or sample size is required, such as comparing a sample mean to a known population mean, or a sample proportion to a known population proportion. More complex calculations are required for studies involving ANOVA, crossover studies, regression, and diagnostic tests.
Everitt BS. The Cambridge Dictionary of Statistics in the Medical Sciences. Cambridge, UK: Cambridge University Press, 1995.
CONSORT Website.
STROBE Website.
Collaboration C. Cochrane Handbook for Systematic Reviews of Interventions, 2011.
Altman DG, Bland JM. How to obtain the confidence interval from a P value. BMJ (Clinical research ed.) 2011;343:d2090.
Altman DG, Bland JM. Uncertainty and sampling error. BMJ (Clinical research ed.) 2014;349:g7064.
Doll H, Carney S. Statistical approaches to uncertainty: P values and confidence intervals unpacked. Equine veterinary journal 2007;39(3):275-6.
Walsh JM, McGowan CA, Mahony RM, Foley ME, McAuliffe FM. Obstetric and metabolic implications of excessive gestational weight gain in pregnancy. Obesity (Silver Spring, Md.) 2014;22(7):1594-600.
Prosser SJ, Miller YD, Thompson R, Redshaw M. Why ‘down under’ is a cut above: a comparison of rates of and reasons for caesarean section in England and Australia. BMC pregnancy and childbirth 2014;14:149.
Researchers often need to evaluate the agreement between two measurements of continuous data (as opposed to categorical data); for example, blood pressure, tumour diameter, forced expiratory volume in one second. This could be measurement of a single feature by two people, or two measurements of the same feature by the same person. Martin Bland and Doug Altman give good advise on how to address this problem. They recommend 1) a graphical method and 2) an arithmetic method, the latter consistent with the British Standards Institution guide for reproducibility of a standard test method. (See: British Standards Institution. Precision of Test Methods 1:
Guide for theDetermination and Reproducibility for a Standard
Test Method (BS 597, Part 1). BSI: London, 1975).
Bland and Altman’s graphical method for assessing agreement is clearly and elegantly described in their 1986 paper ‘ Statistical methods for assessing agreement between two methods of clinical measurement’ Statistical methods for assessing agreement between two methods of clinicalmeasurement (https://www.ncbi.nlm.nih.gov/pubmed/2868172). The paper guides the reader through the process of constructing what is now known as a ‘Bland-Altman Plot’, a plot of the mean of each pair of measurements against the difference of each pair of measurements, which visually identifies the agreement between the pairs of measurements; those outside the ‘limits of agreement’, conventionally within +/- two standard deviations of the mean difference, are instantly apparent. The plot also depicts any bias (for example, does one method of measurement consistently measure higher or lower than the other), and any change in agreement with the magnitude of the measurement, for example, measurements might have good agreement at low values and poorer or more biased agreement at higher values. For further excellent discussion see Watson and Petries’ paper “Method agreement analysis: A review of correct methodology” http://www.theriojournal.com/article/S0093-691X(10)00023-3/abstract and Bland and Altman’s more recent 2003 paper “Applying the right statistics: analysis of measurement studies (https://www.ncbi.nlm.nih.gov/pubmed/12858311).
Study Design
In this section, key elements of systematic reviews and meta-analysis, controlled trials, observational cohort, case-control and cross sectional studies, and case series and case reports, are described.
The most common types of study design are meta-analysis or systematic review, controlled trials, which are ideally randomised controlled trials, the observational study group which includes cohort, case-control and cross sectional studies, case series and case reports, and laboratory or experimental trials. Research projects to develop or compare diagnostic tests usually follow a cohort-type design, although randomised controlled trials are best to see if a diagnostic test improves actual patient outcome. There are a huge number of study designs, most of which are a sub-type of one of these classifications.
This post will give you an overview of study design and advantages and disadvantages of each.
Why bother to understand study design and be able to distinguish between different types? As a clinician, you will use this understanding to:
• critically evaluate reports from the published literature or elsewhere, for example from the media;
• select a suitable study design for your own research project.
The ‘evidence pyramid’ shows the theoretical strength of evidence reflected by various types of study.
It is important to remember that these classifications are a guide, not hard and fast rules. Strength of evidence is governed by the quality of the study, including internal and external validity, as well as the design. A well designed and conducted cohort study probably provides stronger evidence for a given proposition than a poorly conducted randomised controlled trial.
Terminology: exposures and outcomes
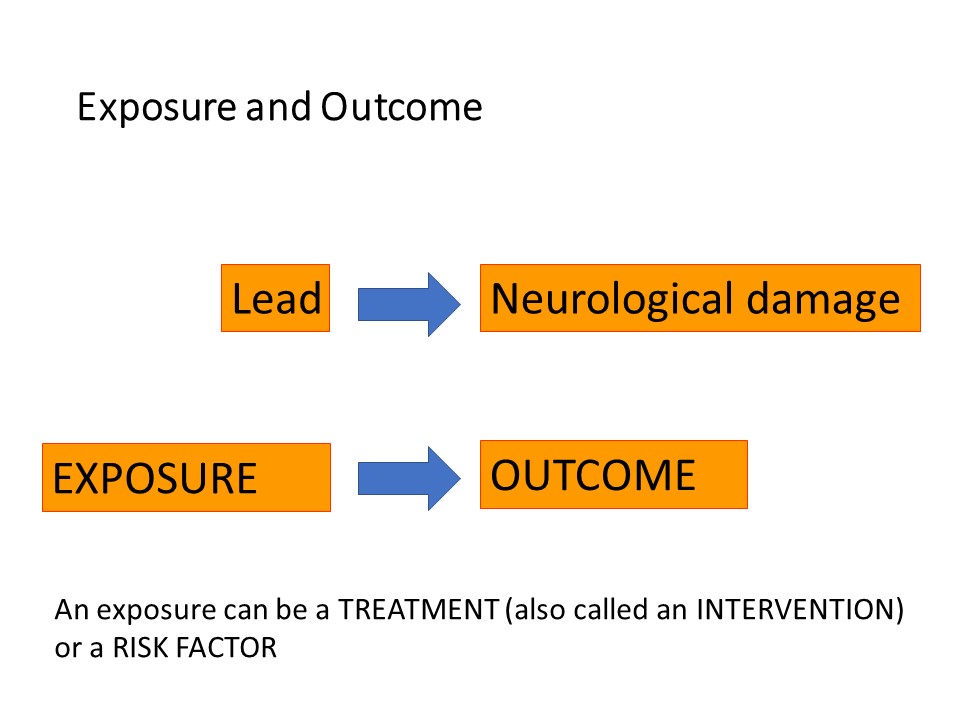
First of all, some background. Epidemiologists and clinical researchers talk about ‘exposures’ and ‘outcomes’. An exposure is the event or risk factor that happens FIRST and causes or influences what happens – the outcome.
An ‘exposure’ can be a medical or surgical treatment, such as a drug or type of surgical procedure. It can be a risk factor, which could be a literal exposure such as exposure to lead or sunlight, or a protective factor such as eating a healthy diet or sunscreen. Factors such as sex or age are exposures. One is ‘exposed’ to being male, or ‘exposed’ to being young. Exposures are also called ‘interventions’, you can understand this in the context of drugs, surgical procedures, diet, physiotherapy and a host of other things which might be recommended to patients.
The ‘outcome’ always comes after the exposure. For example chronic exposure to lead, particularly in children, causes neurological damage, here neurological damage would be the outcome. Eating a healthy diet would protect a person from obesity and cardiovascular disease.
Exposure comes before outcome
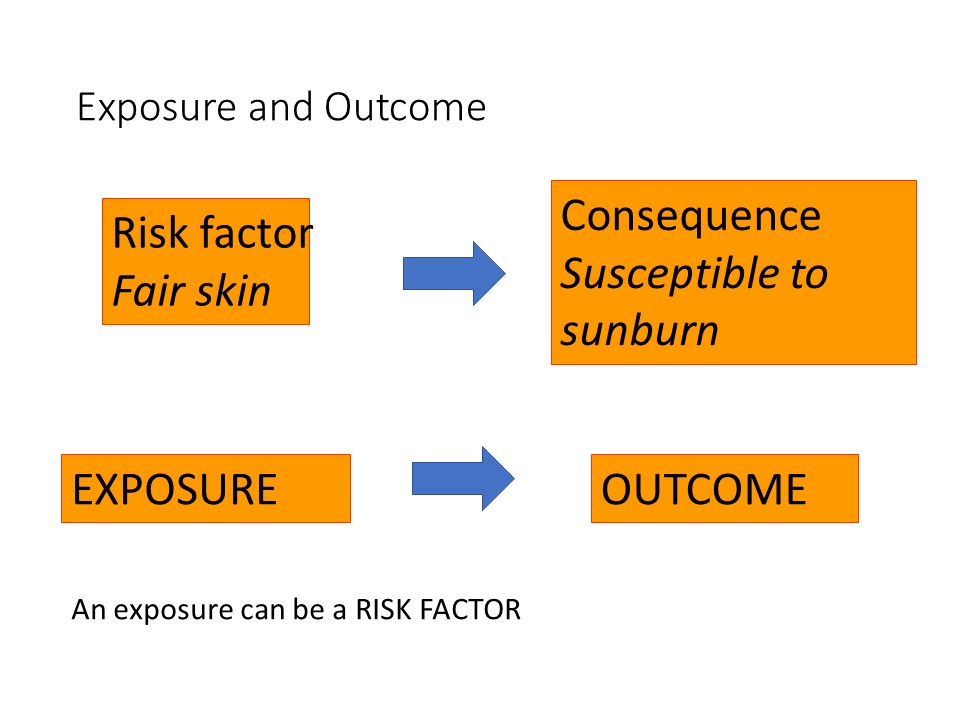
Having fair skin is an example of a risk factor that would make a person susceptible to sunburn.
Fair skin is a risk factor for sunburn
A basic understanding of the classic epidemiological randomised controlled trial and observational study designs are essential to understanding systematic reviews and meta-analysis.
Different types of study depend on how the study participants or subjects are selected, according to the exposure and outcome, and who allocates the study subject to which group. Note that epidemiological study designs are loosely distinguished from those traditionally derived from agricultural studies such as factorial and latin square designs, although there is considerable crossover.
Intervention studies and observational studies
Interventions studies are those in which the researcher allocates some type of intervention or treatment to two or more groups of subjects. The randomised controlled trial is the classic intervention study.
Observational studies are those in which the researcher merely observes the effect of some exposure on two or more groups of subjects. Cohort, case-control and cross sectional studies are the classic observational study designs, often known as ‘epidemiological’ studies.
Randomised controlled trials
In a controlled trial (sometimes known as a clinical trial, although ‘clinical trial’ is not a very precise term), the researcher allocates the study subjects into treatment and control groups.
In order to make a valid comparison of the outcome depending on which treatment a subject is assigned to, the study groups need to be similar at baseline. If the study is not randomised, there is a high probability that the groups will NOT be similar at baseline, i.e. the beginning of the study. Methods of allocating subjects to groups on the basis of, for example, every second patient, or patients in every second week, are not reliable in producing groups which are similar at baseline.
Randomisation
To overcome the problem of creating groups which are similar at baseline, subjects should be randomised to one group or another (usually a treatment and control group). The key point is that each subject must have an equal chance of being allocated to one group or the other. Be suspicious if you read a paper which does not state whether, or how, randomisation was done!
Example of a randomised controlled trial: MRC Trial of streptomycin for tuberculosis 1948
You can find many examples of good randomised controlled trials in your own field of interest. To give you an example of one of the early landmark randomised controlled trials, in 1948 the British Medical Research Council conducted a trial of streptomycin in tuberculosis. The famous epidemiologist and statistician Austin Bradford-Hill was on the comittee and performed the randomisation, where 55 patients were randomly allocated by the researchers to streptomycin plus bed-rest and 52 to bed-rest alone. Patients in the treatment group were given two grams of streptomycin daily intramuscular in four divided doses for four months. All patients received 6 months bed-rest. At the end of the trial only 7% of the treatment group had died, compared with 27% of the bed rest group. This difference in the proportion of deaths was statistically significant at p<0.01.
Key points:
107 patients, 55 allocated to streptomycin plus 6 months bed-rest and 52 to 6 months bed-rest alone
2g streptomycin daily divided: I/M four times daily for 4 months
4/55 (7%) of the streptomcin group vs 14/52 (27%) bed-rest alone group died before the end of 6 months P<0.01
These days we use computerised randomisation. The figure below shows you what a such a randomisation looks like. In this example, each subject in the study (identified by patient number) is randomised to either treatment A or B. Features of randomised controlled trials
Subjects are allocated to groups by the researcher
Valid randomisation technique so that groups are similar at baseline (except for the intervention of interest)
Subjects should not know which treatment they are getting (if they know they are getting a placebo they will expect it ‘not to work’) (Blinding)
Researchers measuring the outcome should not know which treatment the study subject has had (If they know the subject has had the active treatment they might nudge the outcome up to a ‘better’ outcome unconsciously or even consciously (‘Double Blinding’).
Advantages of randomised controlled trials
The ‘gold standard’ for evaluating therapies
Avoid confounding – ‘confounders’ which might have caused the effect instead of the treatment are equally distributed in each group due to randomisation.
Disadvantages of randomised controlled trials
Takes a lot of time and organisation (randomisation, blinding, staff)
Expensive
Unethical where the exposure might cause harm.
Subtypes of randomised controlled trials: parallel and crossover designs
There are many subtypes of randomised controlled trials. The main ones are:
Parallel group – two or more groups run in parallel at the same time – this is the most common design.
Crossover design – In a crossover study, each subject receives both the treatment and control intervention, with each subject acting as their own control. The order or treatment is randomised. Half the subject gets the active intervention first, and the other half get the placebo. After a ‘washout period’, subjects received whichever treatment they did not get during the first ‘arm’. This design is suited to conditions in which the severity of disease does not fluctuate over time, where the treatment is expected to have an effect in a relatively short time and to ‘wear off’ quickly, i.e. have a short duration of action. It is very important that there is an adequate washout period so that the treatment during the first period does not affect the outcome during the second period. In crossover studies, each subject acts as their own control, reducing variation, so sometimes crossover studies can be conducted with fewer subjects than parallel group trials. However analysis of crossover designs can be very complex.
There are many more complex designs based on the idea of random allocation.
Cohort studies
Features of cohort studies
The researcher looks for subjects who receive the exposure but DOES NOT decide who will get the exposure
The researcher then tries to find similar subjects who have not received the exposure
SUBJECTS ARE SELECTED ON EXPOSURE
and a NON-EXPOSED group selected
The outcome is determined after following the subjects over time.
Cohort studies are similar to randomised controlled trials in concept, except that the researcher does not allocate the exposure.
Many of you have seen this figure before, it depicts two groups. The line indicates a timeline at time zero, the start of the study, through to time 2 and beyond. The two groups are followed over time and then the outcome determined.
Example of a cohort study: the British Doctors Study
The British Doctors Study, conducted by the British Medical Research Council during the 1950’s, is the classic cohort study. At the time there was controversy about whether or not smoking was harmful. It would have been unethical to randomise subjects to either smoke or not smoke, so the researchers selected a well-defined group, British doctors, and divided them into a ‘smoking’ and ‘non-smoking’ cohort, and followed them over time. By 1954 it was apparent that smoking was related to a number of adverse outcomes, including premature death, lung cancer, myocardial infarction and respiratory disease.
The reports have been published in 1954, 1956 and 2004.
Advantages of cohort studies
Can calculate relative risk and disease odds ratio
One advantage of a cohort study is that one can infer causation, because the hyothesised ‘cause’ comes temporally before the outcome, i.e. comes before the outcome in time, similar to a randomised controlled trial.
Incidence of disease in exposed and unexposed subjects can be calculated
In prospective studies, data collection is well designed and complete and bias due to faulty recall of events, particularly exposures, is minimised.
It is theoretically possible to undertake a retrospective cohort study, if there are very good records and complete databases available.
Disadvantages of cohort studies
Exposed and unexposed proportions in the target population cannot be estimated
Unsuitable for rare diseases because large numbers of subjects would need to be studied
Potentially long duration of follow-up
Dropouts to follow-up are likely & maintaining follow-up is difficult
Control of extraneous variables may be incomplete
Potentially expensive
Case-control studies
Features of case-control studies
SUBJECTS ARE SELECTED ON DISEASE
and a comparable NON-DISEASED group selected
Cases selected first
Controls selected as close as possible to cases except for disease or outcome of interest
(i.e. controls are ‘matched’ to the cases either as a group or on an individual basis)
‘Look backward’ to see what the exposure was
Example of a case-control study: Helicobacter pylori & gastritis
Barry Marshall, a young resident at the Royal Perth Hospital, and Robyn Warren, a pathologist at the same hospital, were interested in a spiral bacteria they had noticed on endoscopic gastric biopsies of people with gastritis. They hypothesised that these hitherto unknown and unidentified bacteria were associated with gastritis. From a sample of 100 people undergoing gastroscopy for various reasons, they selected those with gastritis and those without gastritis, and compared their exposure to the as yet unclassified organism.
They found a significant association between gastritis and the presence of bacteria in the biopsy specimen (p<0.0001).
This data was published in the Lancet in 1984. The pair, having had their initial abstract rejected at an a local scientific conference in 1983, went on to win a Nobel Prize. See (https://www.nhmrc.gov.au/media/podcasts/2009/conversation-professor-barry-marshall).
Advantages of case-control studies
Good for rare diseases
Good for diseases with long incubation periods
Quick to organise and conduct
Relatively cheap
Require relatively few subjects
Sometimes can use existing records
No risk to subjects
Allow assessment of multiple exposures
Disadvantages of case-control studies
Can’t estimate exposed and unexposed proportions in target population
May be recall bias for exposure
May be hard to validate exposure
Incomplete control of extraneous variables
Difficult to select control group
Incidence of disease in exposed and unexposed subjects can’t be estimated
Cross sectional studies (surveys)
Features of cross sectional studies
In a cross sectional study, A REPRESENTATIVE SAMPLE OF THE POPULATION IS TAKEN
Exposure and outcome are ascertained after data collection
May be cross sectional – undertaken over a short specified time period – a single ‘snapshot’ in time (eg prevalence surveys)
May be longitudinal – observations are repeated over time to provide information about the course of disease over time and space (eg incidence risk or incidence rate)
Example of cross sectional study: oral health in rural children
Ha and colleagues wished to evaluate a number of risk and causative factors related to a number of oral health outcomes in Australian children. They conducted a large survey and then analysed the data for associations between factors such as rurality, indigenous status, being from a fluoridated water area, and socioeconomic status with outcomes such as number of teeth with caries, and decayed, missing or filled deciduous teeth.18
Advantages of cross sectional studies
If a random sample of the target population is selected, can estimate prevalence and proportion of exposed and unexposed subjects in the target population
Quick to conduct
Relatively cheap
Sometimes can use current records
No risk to subjects
Can assess multiple exposures and outcomes
Disadvantages of cross sectional studies
Unsuitable for rare diseases
Unsuitable to diseases of short duration
Hard to control extraneous variables
Can’t estimate incidence in exposed and unexposed individuals
Can’t determine cause and effect (temporal exposure to outcome sequence can’t be evaluated)
Case series and case reports
There are just a few comments on case series and case reports. These type of reports have no control group, so effects of treatment are very unreliable because disease may fluctuate or resolve naturally.
Case series and case reports are very important in flagging the emergence of new diseases and recognition of syndromes. There are many examples of important conditions which were identified initially through case series or case reports, including
HIV/AIDS
Hendra Virus disease
Cystic Fibrosis
Laboratory or experimental studies
In the context of human medicine, laboratory studies or experimental studies are usually like a randomised controlled trial but conducted under artificial, controlled circumstances. There studies are generally conducted in species other than humans, and even for those species, are not ‘real world’ as the animals are husbanded under very artificial conditions and indeed are usually themselves very genetically homogeneous and often genetically modified.
These studies are very important for basic science but are not generalisable directly to clinical practice.
Systematic reviews
In a systematic review, all relevant studies have been systematically identified, appraised and summarised using explicit and reproducible methods (search terms, databases).
Meta-analysis
In a meta-analysis, results from all relevant studies are systematically identified, appraised and summarised using explicit and reproducible methods as for a systematic review. Additionally, the results for each included report are analysed statistically to give an overall summary result.
Example of meta-analysis: streptokinase for acute myocardial infarction.
Joseph Lau and colleagues in 1992 conducted a meta-analysis of mortality after treatment of myocardial infarction with intravenous streptokinase. Such studies had been carried out from 1959 and were still being carried out at the time of publication of the analysis in 1992. The first block represents a conventional meta-analysis, representing nearly 37,000 patients, arranged in order of year of publication from the top. The odds ratio for each individual study is represented as a horizontal line. Lines which do not cross one (the vertical line) are ‘statistically significant’ in their own right. The overall pooled estimate at the lower left shows that streptokinase is highly effective at reducing mortality.
The authors then performed a cumulative meta-analysis, using special techniques, which showed that by the time 2,432 patients had been evaluated, there was a clear advantage of streptokinase; this would have been known by 1973, years before ‘clotbusters’ became routine therapy for myocardial infarction.
One lesson from this is that in many cases, it may be important to do a meta-analysis before conducting a new study – the answer may already be out there!
The focus of our work is to support people who are primarily clinicians – veterinarians, medical doctors and veterinary and human allied health professionals – to undertake research and translate research into practice.
Publication is a key part of the research process, to disseminate your findings beyond your own network. Successful publication starts at the design stage!